Differenzial Expressionen (OMICS) in Excel
Dieses Tutorium zeigt Ihnen, wie Sie eine Differenzial-Expressions-Analyse in Excel mithilfe der Statistiksoftware XLSTAT einrichten und interpretieren.
Datensatz für die Durchführung einer Differenzial-Expressions-Analyse in XLSTAT
Für dieses Tutorium verwenden wir eine simulierte Datentabelle, die 36 biologischen Stichproben kranker und gesunder Individuen entspricht, die zu drei verschiedenen Genotypen gehören. Für jede Stichprobe wird die Expression von 1561 Genen durch RNA-Quantifizierung gemessen. RNAs werden in Zeilen und Stichproben in Spalten gespeichert. Ein Genotypfaktor und ein Gesundheitszustands-Faktor werden rechts in der Datenmatrix eingefügt. Die Zeilennummern der Faktoren entsprechen der Anzahl der Stichproben (Anzahl der Spalten der Datenmatrix).
Die Absicht dieses Tutoriums besteht darin, das Differenzial-Expressions-Tools in XLSTAT zur Identifizierung differenzial exprimierter Gene gemäß zwei Faktoren zu identifizieren: Genotyp (drei Niveaus: BB, BK, KK) und Gesundheitszustand (zwei Niveaus: gesund und krank). Für jeden Faktor werden wir:
- Ein nichtspezifisches Filtrieren durchführen, um Merkmale mit sehr geringer Variabilität zu entfernen.
- Klassische ANOVAs mit einem Faktor automatisch für jedes der restlichen Merkmale durchführen und p-Werte extrahieren.
- p-Werte mithilfe geeigneter Methoden korrigieren, um zu vermeiden, dass signifikante Effekte durch Zufall entdeckt werden. Die mit den niedrigsten p-Werten verbundenen Merkmale (Gene, dargestellt durch RNAs) sind diejenigen, die am signifikantesten von den untersuchten Faktoren beeinflusst werden. Dieses Tool ist sehr hilfreich, um Genpools zu entdecken, die beispielsweise mit einer Krankheit zusammenhängen. Für Faktoren mit mehr als zwei Niveaus (z. B. Genotyp) können wir mehrfache paarweise Vergleiche nach Leistungsmerkmal durchführen. Für Faktoren mit zwei Niveaus (z. B. Gesundheitszustand) können wir Vulkan-Diagramme erzeugen, um sowohl die statistischen als auch biologischen Signifikanzen im Zusammenhang mit allen Merkmalen abzubilden. Beachten Sie, dass das Differenzial-Expressions-Tool in XLSTAT auch zur Untersuchung der Effekte von erklärenden Variablen auf die Proteinproduktion oder Metabolit-Regulierung im Kontext eines hohen Datendurchsatzes bei OMICs verwendet werden kann.
Differenzial-Expression in XLSTAT: Einrichten der Analyse
Um eine Differenzial-Expressions-Analyse durchzuführen, klicken Sie auf XLSTAT-OMICs/Differenzial-Expression. Wählen Sie in der Registerkarte Allgemein die Datenmatrix im Tabellenfeld Merkmale/Individuen aus. Hier werden die Individuen durch unsere Stichproben repräsentiert. Sie müssen die Option Merkmale in Zeilen nicht ändern, da die Gene im Datensatz in Zeilen gespeichert sind. Die erste Spalte im Datensatz, die die Leistungsmerkmal-IDs enthält, muss unbedingt ausgewählt werden. XLSTAT benötigt diese Informationen, damit der Benutzer interessante Merkmale mit ihren Namen in der Analyseausgabe identifizieren kann. Im Feld der erklärenden Variablen wählen Sie die beiden Spalten aus, die die Zugehörigkeit jeder Stichprobe zu Faktorniveaus enthält.
Wählen Sie in der Registerkarte Optionen einen Parametrischen Testtyp aus. Mit dieser Option erhalten Sie eine ANOVA mit einem Faktor pro Faktor und Leistungsmerkmal. Bei einer kleinen Anzahl von Stichproben empfehlen wir anstatt dessen die Verwendung der nichtparametrischen Methode, bei der die ANOVAs mit einem Faktor durch Kruskal-Wallis-Tests ersetzt werden. Wählen Sie in den nachträglichen Korrekturen das Benjamini-Hochberg-Verfahren, das bei Differenzial-Expressions-Studien sehr häufig verwendet wird. Es gehört zur Familie der False Discovery Rate ( FDR ) p-Wert-Korrekturen. Es eignet sich gut für Studien, die die Berechnung einer großen Zahl von p-Werten umfassen, da es weniger stringent ist als Korrekturen, die zu Familie Wise Error Rate ( FWER ) gehören, wie z. B. die Bonferroni-Anpassung. Stellen Sie die Anzahl der niedrigen zu behaltenden p-Werte auf 30 ein, um zu vermeiden, dass riesige Listen mit p-Werten in der Ausgabe angezeigt werden (hohe p-Werte sind im Kontext unserer Studie nicht sehr interessant). Aktivieren Sie die Option mehrfache paarweise Vergleiche und wählen Sie Tukey (HSD), um mehrfache paarweise Vergleiche unter den Genotyp-Niveaus für jedes Gen zu erhalten. Schließlich aktivieren Sie die Option nichtspezifisches Filtrieren, wählen Sie % (Std. Abw.) mit einem Schwellwert von 50 %, um 50 % der Gene basierend auf dem Kriterium der niedrigsten Standardabweichungen vor den Analysen zu eliminieren.

In der Registerkarte Diagramme aktivieren Sie die sowohl die Option Histogramm der p-Werte als auch die Option Vulkan-Diagramm.
Die beiden folgenden Optionen stellen zwei Möglichkeiten der Darstellung biologischer Effekte auf die X-Achse des Vulkan-Diagramms dar. Wir wählen das Log2(Verhältnis der Mittelwerte), da unsere Daten nicht transformiert werden. Aktivieren Sie die Option Merkmale identifizieren. XLSTAT verwendet daher eine spezielle Farbe für hoch signifikante Merkmale sowohl in der statistischen als auch in der biologischen Skala und entsprechend den beiden folgenden Schwellwerten. Wählen Sie 1 für Schwellwert(x). Ein Log2(Verhältnis der Mittelwerte) von 1 bedeutet, dass der Mittelwert im Zähler doppelt so hoch ist wie der Mittelwert im Nenner. Umgekehrt bedeutet ein Log2(Verhältnis der Mittelwerte) von -1, dass der Mittelwert im Nenner doppelt so hoch ist wie der Mittelwert im Zähler. Ein Log2(Verhältnis der Mittelwerte) von 2 oder -2 repräsentiert eine mehrfache Veränderung von 2², usw. Wählen Sie einen p-Wert-Schwellwert von 0,001 im Feld Schwellwert(y). Das bedeutet, dass der Schwellwert für die statistische Signifikanz –log10(0,001) ist.
Klicken Sie auf den Button OK.
Differenzial-Expression in XLSTAT: Interpretieren der Ergebnisse
Nach einer Zusammenfassung zu verschiedenen Optionen, die in der Analyse verwendet wurden, wird die Anzahl der Merkmale, die durch das nichtspezifische Filtrieren eliminiert wurden, angezeigt. Dann wird eine Analyse pro Faktor angezeigt.
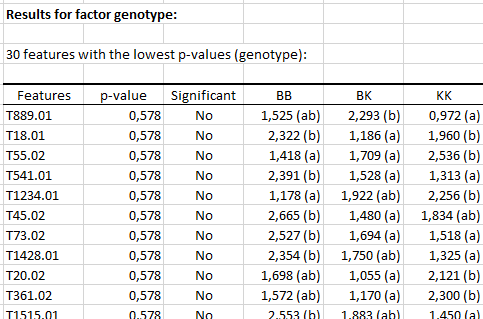
Zuerst wird eine Tabelle mit den 30 signifikantesten Merkmalen angezeigt, die nach steigenden p-Werten sortiert sind. Die Tabelle enthält den Namen des Leistungsmerkmals, penalisierte p-Werte, die Signifikanz und die RNA-Quantitätsmittelwerte für jedes Faktorniveau. Wenn ein p-Wert signifikant ist, ist der Benutzer möglicherweise an mehrfachen paarweisen Vergleichen interessiert, die durch mit Mittelwerten verbundene Buchstaben dargestellt werden. Zwei Niveaus mit demselben Buchstaben sind nicht signifikant verschieden. Zwei Niveaus, die keinen Buchstaben gemeinsam haben, unterscheiden sich signifikant.
Für den Genotyp-Faktor gibt es keinen signifikanten p-Wert für alpha = 0,05. In diesem Fall ist die Interpretation von vielfachen Vergleichen für keines der Merkmale relevant.
Das p-Wert-Histogramm zeigt, dass die p-Werte homogen verteilt sind.

Der Faktor gesund oder krank wirkt sich scheinbar auf die Expression von zwei Genen aus: T1157.01 und T106.02. Das erste hat eine höhere Expression bei gesunden Stichproben, und das zweite hat eine höhere Expression in den kranken Stichproben.

Beide Merkmale können im Vulkan-Diagramm dargestellt werden:

Merkmale, die sich oben links und oben rechts des Diagramms befinden, werden beschriftet. Sie entsprechen den Merkmalen, die die biologischen und statistischen Signifikanz-Schwellwerte (gestrichelte Linien) überschreiten.
Beachten Sie, dass die p-Werte, die im Vulkan-Diagramm zum Berechnen der –log10(p-Werte) verwenden werden, die rohen, unkorrigierten p-Werte sind.
War dieser Artikel nützlich?
- Ja
- Nein